Proper Structure of Amino Acids You Need to Know for the Mcat
Learn primal MCAT concepts near proteins, the structure and classification of amino acids, plus practice questions and answers
(Note: This guide is role of our MCAT Biochemistry series.)
Part 1: Introduction to proteins
Function two: Amino acids
a) Structure of amino acids
b) Classifying amino acids
Role three: Peptide bonds
a) Formation
b) Hydrolysis
Office 4: Protein structure
a) Primary, secondary, tertiary, and fourth structures
b) Specialized amino acids
c) Stability and interactions
d) Protein folding
Part 5: High-yield terms
Part 6: Passage-based questions and answers
Office 7: Standalone questions and answers
Part i: Introduction to proteins
Proteins are an incredibly high-yield concept on the MCAT, simply like a lot of biochemistry topics, they aren't easily mastered without a great deal of practice. These topics are specially intimidating considering there is virtually no limit to what you can learn about proteins, amino acids, and everything else.
This guide will serve as an introduction to amino acids, protein structure, and protein interactions. While it volition non be a comprehensive handbook to everything about proteins, it will exist a skilful identify to beginning studying these basic principles of biochemistry. Be sure to refer to our other biochemistry guides for further information on proteins, enzymes, and other biological molecules.
Throughout the guide, you will encounter several bolded terms. Their definitions are peculiarly important and can likewise be institute in Part 4 of this guide. At the end of this guide, you will also find several passage-based and standalone questions to sharpen your skills.
Let's begin!
Part 2: Amino acids
a) Structure of amino acids
Amino acids are the edifice blocks of all proteins. The structures of amino acids are an extremely high-yield topic to report.
The structure of each amino acid tin can exist divided into iii separate regions:
-
The amino group, or N-terminus
-
The carboxylic acid group, or C-terminus
-
A unique identifying side chain, or R-grouping

Effigy: Each amino acid has an amino grouping, an R grouping, and a carboxylic acid grouping.
Recall that an amino group is a functional group composed of NH3+. Information technology is similar to ammonia (NH4), except that at one position, the nitrogen is attached to a carbon instead of a hydrogen (NH2C instead of NH3). Note that this results in one free electron pair on the nitrogen cantlet. At physiological pH (pH ~7), this free electron pair is able to accept a bail to a single hydrogen atom. This results in a positive charge on the functional group.
The acid on every amino acid is a carboxylic acid, a functional group composed of COOH. At physiological pH (pH ~7), this carboxylic acid is deprotonated, leaving a negative charge on the functional group.
Note that at physiological pH, amino acids are zwitterions; they contain both positive and negative charges on the same molecule. Most amino acids have a net charge of zippo. Exceptions arise when accounting for charges on the R-grouping, or side chain, of the amino acid.
These R groups, or side chains, can be as simple equally a single hydrogen atom or equally complex as an imidazole ring. In that location are 20 different R groups—each of which y'all should commit to memory. We'll hash out these side bondage farther in the adjacent section.
The R group is connected to the central carbon, which is known as the alpha carbon. This carbon is connected to every constituent of the amino acid: the amino grouping (-NH3+), the carboxylic acid part (-COO-), the R grouping, and a hydrogen atom (H).
Annotation that for xix of the twenty amino acids, the alpha carbon itself is chiral, or attached to four different constituent groups. (The exception happens to exist glycine, as the R grouping is simply a hydrogen cantlet.) Chirality refers to right- or left-handedness, denoted as D- and L- molecules, respectively. The chirality of biological molecules becomes quite important, as only 50-configuration (left-handed) amino acids can be used past the torso. (D-amino acids are non naturally institute in eukaryotic metabolic pathways.)
b) Classifying amino acids
Each amino acid has a characteristic side chain, and the properties of these side chains are essential for the office of proteins.

Figure: A table of amino acids, including 3- and ane-letter abbreviations, side chains (highlighted), and the pKa of any acidic or basic side chains.
Notation that there are three ways to refer to an amino acid: by its full proper name, its three-letter abridgement, or its one-letter abridgement. The MCAT may exam your cognition of all three, so be certain to memorize each form.
Nonpolar side chains
At that place are viii nonpolar amino acids: alanine, phenylalanine, valine, leucine, isoleucine, tyrosine, tryptophan, and methionine. These side chains are considered nonpolar due to their aliphatic nature; they are primarily composed of carbons and hydrogens.
Due to their nonpolar nature, these amino acids will not be attracted to water surrounding the protein. Nonpolar amino acids are frequently located in the core of proteins (proteins are 3-dimensional) in aqueous solution. These nonpolar amino acids are essential to forming the construction of the protein.
Polar side chains
At that place are 4 polar amino acids: serine, threonine, asparagine, and glutamine. Due to the attraction between water and other polar molecules, these amino acids are often positioned on the exterior of the poly peptide or the protein's agile site. Polar side bondage volition exist attracted to other polar side bondage and to water molecules in the aqueous solution that surrounds the poly peptide.
Acidic side chains
There are two acidic amino acids: aspartate and glutamate. These residues are proton donors. They will frequently lose a proton in solution becoming negatively charged.
While studying for the MCAT, it's worth noting the pKa of these acidic side chains besides. Yous should be able to recognize if these side chains should be protonated or deprotonated in any given range of pH values. Recall that pKa is the pH at which one-half of the group (or molecule) of interest is deprotonated and half is protonated. Thus, at pH=three.7, roughly half of the aspartate side bondage within a protein will be deprotonated, while the other one-half is protonated. Thus, at pH ~ seven, most aspartates are deprotonated (and negatively charged).
Basic side chains
At that place are three bones amino acids: lysine, arginine, and histidine. These residues are proton acceptors. Each of these basic amino acids has a nitrogen that can accept a hydrogen to become positively charged.
As with acidic amino acids, yous should also be familiar with the pKa of each of these side chains and be able to recognize if they are protonated or deprotonated within a given range of pH values.
Note that although tyrosine possesses an -OH group at the end of its side chain, its hydroxyl group is rarely constitute deprotonated at whatever physiologically relevant pH. Thus, information technology is considered a nonpolar amino acid--rather than an acidic one.
Function 3: Peptide bonds
Proteins are composed of amino acids bound together through peptide bonds. The formation of the peptide bond is catalyzed past the ribosome. As the ribosome reads an mRNA strand, information technology translates and adds amino acids to the growing polypeptide. You can read more about protein synthesis in our guide on RNA.
a) Formation
The formation of a peptide bail between two amino acids, or between an amino acid and a peptide, is an instance of a nucleophilic exchange reaction (a subset of nucleophile-electrophile reaction)—a very common reaction that you should know for the MCAT.
The reaction machinery is drawn below.

Figure: The nucleophilic substitution reaction leading to formation of a peptide bail.
Notation that the carbonyl grouping of the carboxylic acid is an electrophile. The nucleophile is the nitrogen of the amino group. The nitrogen of the 2nd amino acrid has a gratuitous lone pair that tin can attack the carbonyl grouping, forming an Due north-C bond.
In the procedure, electrons from the second bond of the C=O are sent to the carbonyl oxygen. These electrons then reform a 2d bond, and the leaving group (-OH) leaves with a lone pair of electrons to grade a molecule of h2o. This reaction tin too be referred to every bit a dehydration reaction.
A peptide bond is formed between the nitrogen of the amino group of an amino acid and the carbon of the acid group (carboxyl) of another amino acid or growing peptide strand). This bail is an amide bond. The carbon of the amide bail is too double-bonded to an oxygen atom. Considering the nitrogen of the peptide bail has a alone pair, the peptide bond has a fractional double bail graphic symbol.

Effigy: Resonance structures of the planar amide bond.
Remember that while unmarried bonds tin freely rotate, double bonds cannot. Since the peptide bond has a partial double bond grapheme (think of information technology as an average of a single and double bail), it does non rotate as much as a single bond and tin can exist treated as a fixed bond.
b) Hydrolysis
Let's now talk over the reverse reaction, peptide bail hydrolysis. Hydrolysis ways breaking (lysis) with water (hydro). Then, peptide bonds can exist broken past water molecules.

Figure: Peptide bond hydrolysis.
When h2o (or more than specifically a lonely pair on the oxygen of h2o) attacks the carbonyl carbon, the electrons in the pi bond (double bail) move onto the oxygen atom. The nitrogen of the amide bond will leave, and the newly formed amino group volition be protonated by hydrogen atoms that are in the solution.
Afterward a hydrolysis reaction, nosotros are left with two newly formed segments with completed amino and carboxylic acrid groups on either amino acid.
Part 4: Protein structure
a) Primary, secondary, tertiary, and quarternary structures
Proteins are composed of many amino acids linked together through peptide bonds. Before discussing structure, it is important to set some nomenclature. Below is a unproblematic representation of a protein composed of n+ii amino acids.
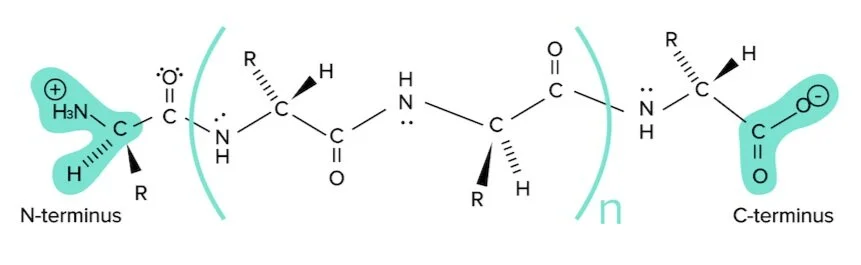
Figure: A unproblematic polypeptide sequence.
Notice the protein is fatigued from left to correct, starting with the N-terminus and ending with the C-terminus. The N-terminus refers to the side of this string with the amino group (or nitrogen) exposed. The C-terminus refers to the side of this string with the carboxylic acid group (or carbon) exposed. This is the writing convention for all protein sequences.
There are four levels of structure: primary, secondary, 3rd, and fourth. They are all critical to the protein's part. The starting time level of protein construction is its primary structure. Principal structure refers to the cord of amino acids connected past peptide bonds and is defined solely past the identity of amino acids within information technology.
Secondary structure is formed through the hydrogen bonding interactions between atoms forming the backbone of the protein chain—rather than interactions between the side chains of each amino acid. Recollect that each amino acrid contains:
-
An N-H group, from the amide bail
-
A C=O bond, from the carboxylic acrid
Secondary structure is composed of the hydrogen bonding interactions between the H of the N-H of 1 amino acid and the carbonyl oxygen (through 1 of its alone pairs) of another amino acid.
There are two principal secondary structures: blastoff helices and beta sheets. The alpha helix is stable because of the many hydrogen bonds that are formed when the courage is arranged in this way. Find that the R groups do not contribute to the hydrogen bonding forming the alpha helix.
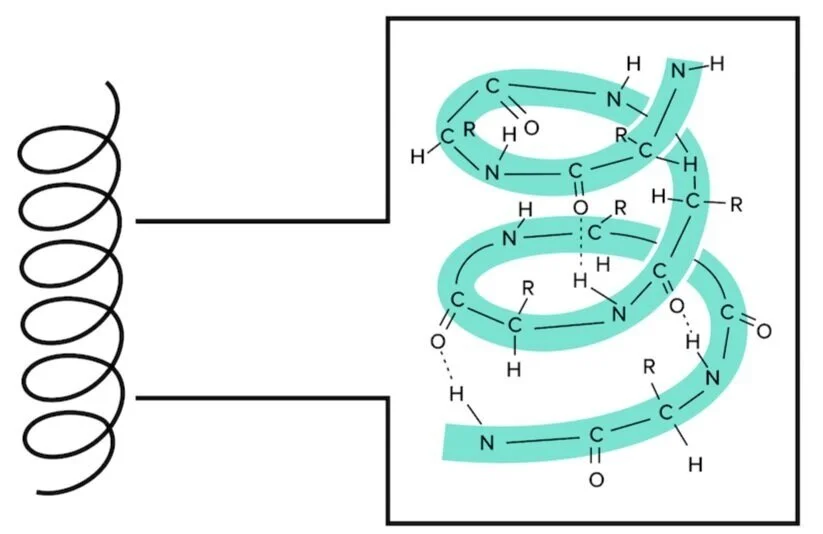
Effigy: An alpha helix is an extremely stable secondary structure.
Blastoff helices serve a lot of different functions in different proteins. Many transmembrane proteins use alpha helices that span the entire membrane to transport ions from exterior to within the protein. You lot can find more information about transmembrane proteins in our guide on Lipids and Membranes.
Beta sheets are also formed through hydrogen bond interactions. However, instead of a helix system, different regions on the amino acid string line upwardly in rows.
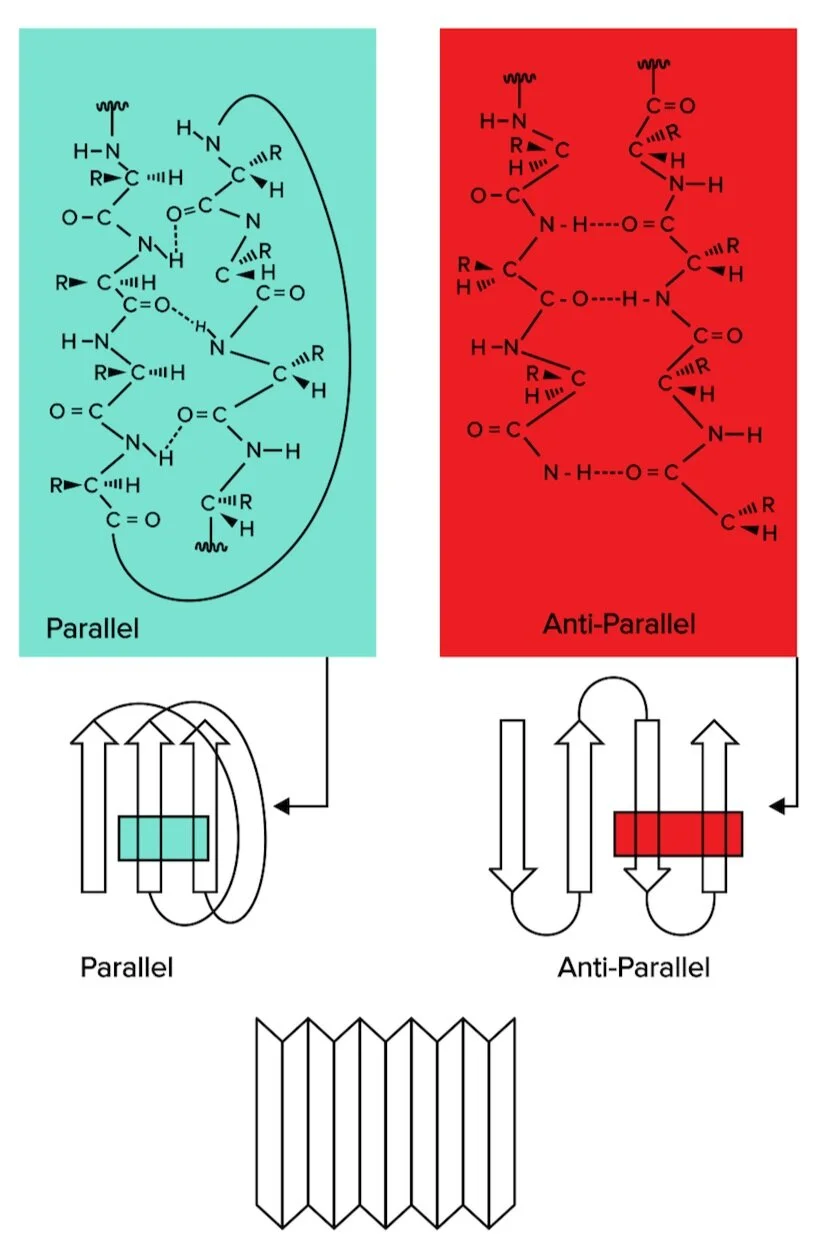
Figure: Beta sheets, another form of secondary structure, can be classified as parallel or antiparallel.
At that place are two types of beta sheets: parallel and antiparallel sheets. In parallel sheets, both strands of the polypeptide chain run in the same direction. In antiparallel sheets, the next strands are running in the opposite directions (1 is going from N to C terminus, while the other runs from C to N terminus).
Tertiary structure refers to structure that arises from interactions betwixt the side chains of different amino acids. Positively charged side bondage (histidine, lysine, and arginine) can collaborate with negatively charged side chains (aspartate, glutamate). Polar side chains volition be attracted to other polar side chains. Nonpolar side chains will exist attracted to other nonpolar side chains. Finally, two cysteine residues tin can covalently bond to each other, forming a disulfide bond.
4th structure refers to interactions between ii unlike poly peptide subunits that make up a protein with more than than i subunit. Many proteins have more than one subunit. Many proteins have more than ne subunit, or polypeptide strands. For instance, there are iv subunits required to form hemoglobin. These interactions can be between backbone or between side bondage. You will often discover covalent disulfide bonds formed betwixt two cysteine residues contributing to 4th construction.
These bonds are formed after the protein has been completely translated from RNA. Such modifications are referred to as post-translational modifications and can include changes such as:
-
phosphorylation at certain amino acrid residues (such as serine, threonine, and tyrosine)
-
formation of disulfide bonds betwixt cysteine residues
-
glycosylation, or the linkage of curt carbohydrate polymers to the protein
-
ubiquitination, or the attaching of a protein known as ubiquitin that marks the poly peptide as a target for degradation
b) Specialized amino acids
Glycine
Think that glycine possesses a single hydrogen as its side chain. Due to the relatively pocket-size size of this side concatenation, glycine is the least sterically hindered amino acid. Every bit a outcome, it tin rotate and move more easily. It is often plant in areas of the protein which need a high corporeality of flexibility and rotation.
Cysteine
Remember that cysteine has a thiol group (Due south-H) at the stop of the side chain. This gives cysteine the ability to bind to other cysteines through the formation of a disulfide bond (S-S). These disulfide bonds are important to both the tertiary and quaternary structures of many proteins. In detail, many proteins with multiple subunits grade bonds betwixt subunits using disulfide bonds.
Proline
In dissimilarity to glycine, proline is the Almost sterically strained amino acid. Annotation that the side chain of proline is jump to the amino group of the amino acid. This creates a five-membered ring that does not take a lot of rotational power and oftentimes creates a kink in the protein strand. This amino acid is often institute in regions of the protein that should exist immobile or when a bend in the structure is needed.
c) Stability and interactions
The multiple levels of structure within a protein are essential for the protein to role properly, especially in the aqueous environment of the human body. Within this environment, the hydrophobic event is an important force to consider.
The hydrophobic effect is a consequence of nonpolar and polar interactions. In an aqueous environment, hydrophobic residues will be attracted to each other, while they will also exist repelled by the polar aqueous environment (water and ions in the water). For this reason, hydrophobic amino acids will be constitute in the interior of the protein rather than the outside of the protein.
This can also be explained from a thermodynamic standpoint. The hydrophobic effect also has to exercise with entropy. Most simply, entropy is a measure of the disorder of a organisation. Systems e'er increment in entropy, resulting in more than disorder with time. If a protein has hydrophobic residues on the outside of the poly peptide, then surrounding water molecules volition have fewer residues to bind to on the surface of the protein.

Effigy: H2o molecules surrounding the protein are known as the solvation/hydration layer.
When the outer protein residues are polar, the water molecules in the solvation layer tin easily grade hydrogen bonds in different rearrangements—h2o molecules in the solvation layer can hydrogen bail with polar residues and the other h2o molecules surrounding information technology.
However, if the outer layer of the protein is composed of mainly hydrophobic residues, the number of hydrogen bonds that the solvation layer can form becomes extremely express. This requires the water molecules to be oriented more precisely/rigidly, resulting in an unfavorable reduction in entropy.
d) Poly peptide folding
The solvation environment of a polypeptide concatenation directly influences the way a protein folds. Protein folding refers to the process through which a protein is organized (or folded) into its proper secondary and tertiary structures. Denaturing refers to the process through which a protein is unfolded or loses its proper 3D construction.
Protein folding can be disrupted by several environmental weather condition, including:
-
Loftier or low pH
-
High table salt concentration (including the presence of molecules like urea)
-
High temperature
These same environmental changes can result in protein denaturation. As protein form is highly related to function, protein denaturation well-nigh oftentimes results in loss of function.
Denaturing a protein disrupts secondary, 3rd, and fourth structures. Notation that protein denaturation does non disrupt poly peptide structure. In some proteins, it may be possible for a protein to refold back to its native state and regain its proper secondary, tertiary, and quaternary structure.
Yet, many proteins cannot refold to the native state once unfolded. Due to the high numbers of possible conformations that the polypeptide sequence can presume, folding a protein can exist a tricky business. In biological systems, proteins chosen chaperones assist in folding denatured proteins back into their native state.
Acknowledgements: Joshua Perez-Cruet
Role five: High-yield terms
Amino grouping: a functional group equanimous of NH3+
Carboxylic acrid: a functional group composed of COOH
Zwitterions: molecules that comprise both positive and negative charges on the aforementioned molecule
Blastoff carbon: central carbon atom of an amino acrid, bonded to the amino group, carboxyl grouping, and R grouping
Peptide bond: bond between each amino acid in a poly peptide; catalyzed past the ribosome; forms primary structure of a protein
Amide bond: some other term for peptide bond; has partial double bail character which limits rotation of the constituent groups
Chief structure: the string of amino acids connected past peptide bonds, and is divers solely by the identity of amino acids within it
Secondary structure: formed through the hydrogen bonding interactions betwixt atoms forming the backbone of the protein chain; includes alpha helices and beta sheets
Tertiary structure: structure that arises from interactions between the side chains of dissimilar amino acids
4th structure: interactions betwixt dissimilar protein subunits that make up a protein with more than one subunit
Hydrophobic issue: a issue of nonpolar and polar interactions; in an aqueous surroundings, hydrophobic residues will be attracted to each other, while they volition also exist repelled past the polar aqueous environment
Protein folding: the process through which a poly peptide is organized (or folded) into its proper secondary and tertiary structures
Denaturing: the procedure through which a protein is unfolded or loses its proper 3D structure
Chaperones: helper proteins that assist in folding denatured proteins back into their native country
Function half-dozen: Passage-based questions and answers

Figure one: Mouse antibody and human antibodies.
Researchers inject mice with an antigen to induce the production of specific antibodies. Upon confirmation of antibiotic production, antibodies are isolated from blood samples taken from the mice and tested to determine if they are specific to the antigen using an enzyme-linked immunosorbent assay. Researchers also cohabit drugs to antibodies to produce drug conjugates called nanobodies. The nanobodies localize to antigens and often take a cleavable linker betwixt the nanobody and drug, with an intention to localize nanobodies to the peripheral membrane poly peptide. Once the nanobody localizes, it can enter the cell through endocytosis and then can be cleaved in a peroxisome or lysosome. Upon cleavage, the drug payload is released. Toxic drugs can be localized to cancer cells without causing undue damage to neighboring cells.
In a follow-upwardly experiment, a nanobody identified as N96 successfully binds to HER2: an epidermal growth gene highly expressed in some chest cancers. Scientists accept assayed the pH dependency of the N96's analogousness for HER2.

Figure ii: pH dependency of nanobody binding.
Question 1: Which of the following is likely to be found in homo antibodies only non mouse nanobodies?
A) An antigen-specific variable region
B) Hydrophobic residues
C) Fourth structure
D) Hydrogen bonding
Question 2: Researchers promise to identify the antigen-binding region of the nanobody. Based on the information provided in Effigy two, which of the post-obit amino acids is most likely in this region?
A) His
B) Arg
C) Lys
D) Asp
Question three: Which of these residues is most likely to exist found on the outside of N96?
A) Threonine
B) Phenylalanine
C) Leucine
D) Valine
Question 4: During drug delivery, the nanobody linker is broken by an intracellular protease that recognizes the pattern X-X-K-N-X-X, where X is any nonpolar amino acid residue. Which of the following amino acids should non exist included in the linker?
A) Isoleucine
B) Valine
C) Glutamine
D) Phenylalanine
Answers to passage-based questions
-
Answer option C is correct. Effigy 1 provides a comparison of the structural differences between mouse and human antibodies. Mouse antibodies are equanimous of a unmarried protein domain produced from a single mRNA transcript. These single subunits are functional on their own, without forming bonds to multiple subunits or creating a 4th structure (option C is correct). Both antibodies and nanobodies accept antigen-specific variable regions (selection A is incorrect). All proteins are probable to have some amount of hydrophobic residues (choice B is incorrect). Hydrogen bonding is required to course secondary and tertiary structures, and thus provide protein function (selection D is incorrect).
-
Reply option A is correct. According to Figure 2, nanobody binding peaks at pH=7-8. Its affinity gradually decreases from pH viii-12 and decreases very quickly from pH 5-6. This rapid decrease in affinity from pH 5-6 is probable due to protonation of some residuum. Recollect that the pKa of histidine is approximately 6, resulting in protonation at this pH level (choice A is right). Both arginine and lysine are basic residues and have higher pKa values (choices B and C are wrong). Aspartate, an acidic residue, has pKa~iii and so would likely be deprotonated throughout this entire range of values (choice D is incorrect).
-
Answer choice A is correct. Threonine is a polar rest containing a hydroxyl (-OH) group (choice A is correct). Phenylalanine, leucine, and valine side chains contain nonpolar residues more than frequently found folded on the interior of a protein (choices B, C, and D are incorrect).
-
Reply choice C is right. The linker can include whatever nonpolar amino acids at the X positions. Isoleucine, valine, and phenylalanine are nonpolar residues that meet these criteria (choices A, B, and D are incorrect). Glutamine is a polar amino acid, so information technology cannot occupy the Ten positions (choice C is right).
Part 7: Standalone questions and answers
Question 1: Which of the following amino acids is naturally found in the R-configuration?
A) Tryptophan
B) Cysteine
C) Tyrosine
D) Methionine
Question 2: Why are hydrophobic residues often found on the interior of a protein?
A) They lower the entropy of the system
B) They are often less bulky than hydrophilic residues
C) Their van der Waals interactions are stronger than hydrogen bonds
D) The solvation layer is less ordered near hydrophilic residues
Question iii: Which of these is a form of primary construction interaction?
A) Disulfide bond
B) Peptide bond
C) Interactions between Due north-H and C=O of the protein backbone
D) Hydrophobic interactions
Question iv: Researchers hope to blueprint a polypeptide inhibitor to a site with the repeated sequence A-Q-Due east-M-Chiliad. Which of these inhibitor sequences is virtually likely to succeed?
A) V-Q-R-A-A
B) Eastward-Q-Due east-K-Grand
C) Due east-Due south-W-D-D
D) A-S-K-D-E
Question 5: Which of the following best describes the issue of peptide bond hydrolysis?
A) The amino group of one amino acid attacks the carboxyl group of another amino acid
B) A water molecule is released into solution
C) A carboxylate group and an amine group are produced from an amide group
D) The protein has been denatured
Answers to standalone questions
-
Answer option B is right. R- and S-configuration refer to the chirality at the alpha carbon of the amino acid. Due to the priority rankings of all side bondage, all 19 chiral amino acids are of the S-configuration except cysteine. This is due to the higher priority of the thiol side concatenation on the cysteine molecule, making it an R-configuration (choice B is correct). All other amino acids except glycine (which is achiral) are constitute in the S-configuration (choices A, C, and D are incorrect).
-
Answer selection D is correct. From a thermodynamic standpoint, the solvation layer is more ordered when nearby hydrophobic residues are nearby because there are fewer possible ways to create hydrogen bonds (choice D is right). Hydrogen bonds are stronger than van der Waals interactions, and nonpolar residues cannot participate in hydrogen bonding (choice C is incorrect). Localization of hydrophobic residues has less to practice with bulkiness than entropy; in fact, some hydrophilic chains such every bit aspartate and glutamate are quite bulky (choice B is incorrect). Hydrophobic residues lower the entropy of the system when on the exterior of the protein rather than the interior (choice A is incorrect).
-
Answer choice B is correct. Peptide bonds are formed between amino acids, and so comprise the principal structure of a protein (option B is correct). Interactions between Northward-H and C=O groups of the protein backbone class blastoff helices and beta sheets, forms of secondary structure (choice C is wrong). Tertiary construction gives shape to globular proteins and arises from interactions between side chains of amino acids—including hydrophobic side chains (selection D is incorrect). Disulfide bonds between cysteine residues are found contributing to both tertiary structure and quaternary structure (choice A is incorrect).
-
Answer choice D is right. To maximize attraction betwixt the inhibitor and the site, the two sequences must exhibit similar attractions. The catalytic site, with a repeated sequence of alanine-glutamine-glutamate-lysine-lysine, has nonpolar-polar-acidic-bones-basic residues. As a result, the desired inhibitor should accept: nonpolar-polar-basic-acidic-acidic residues to maximize hydrophobic interactions betwixt nonpolar residues and ionic attractions betwixt acidic and basic residues (option D is correct). Placing alanine in the 4th and 5th positions would result in weak interactions with lysine (choice A is incorrect). Placing an acidic residual at position 1 would effect in weak interactions with nonpolar alanine (pick B is incorrect). Tryptophan is not a charged residue, then would not be paired well with glutamate (selection C is incorrect).
-
Answer choice C is correct. Peptide bonds are formed through nucleophilic attack (choice A is incorrect). Peptide bail germination as well results in dehydration, or the release of one water molecule (choice B is incorrect). Denaturation occurs when secondary, tertiary, and quaternary structures are disrupted rather than primary construction (pick D is incorrect).
Source: https://www.shemmassianconsulting.com/blog/proteins-mcat
0 Response to "Proper Structure of Amino Acids You Need to Know for the Mcat"
Publicar un comentario